Introduction to Pathways Project
Lecture is designed to introduce students to the big picture of the Pathways Project.
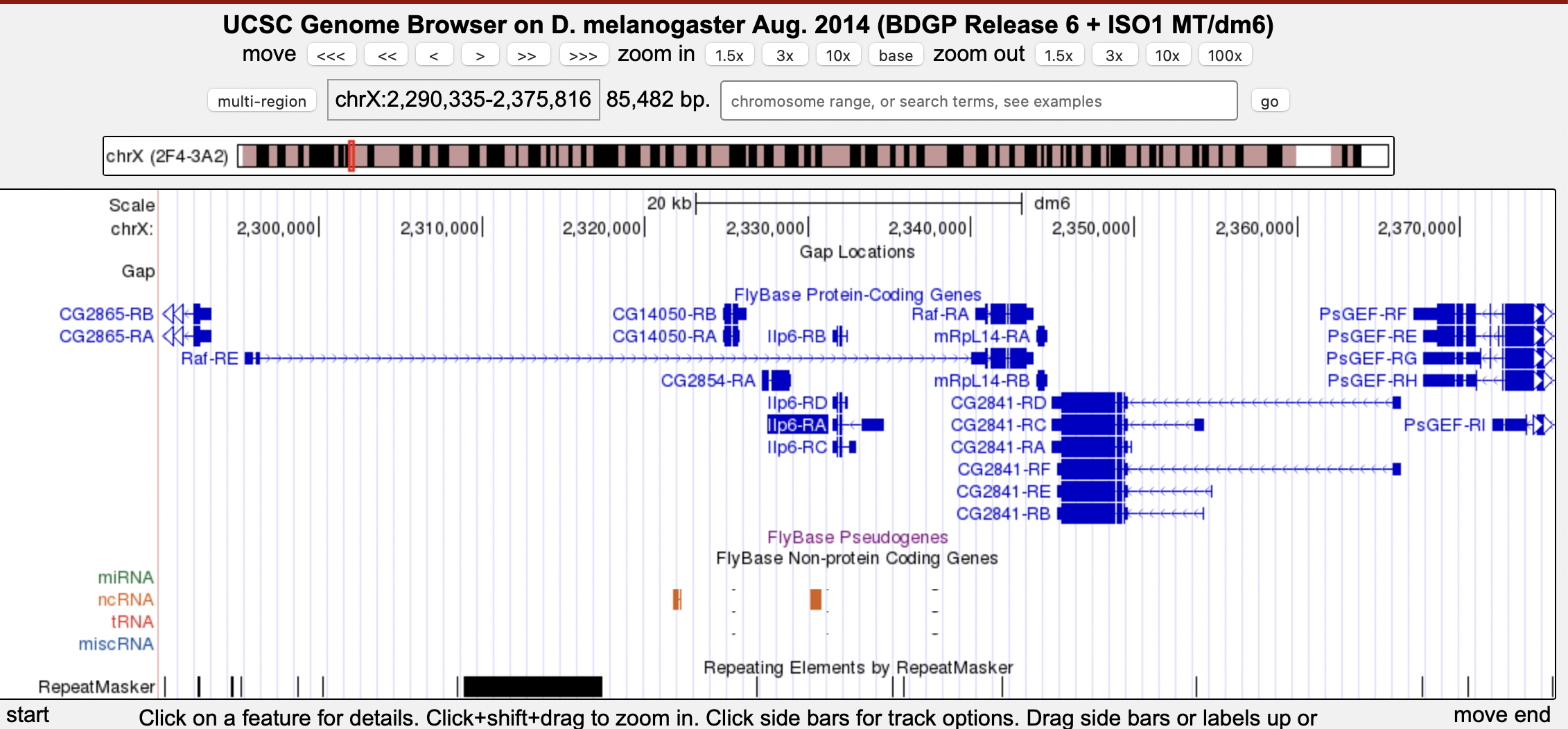
In this example, Ilp6 is within the intron of Raf-PE, however Raf-PA is upstream of Ilp6.
We are defining gene order based on the first/closest protein coding exon only. So if the gene is nested in an intron that is between two non-coding exons then we ignore those UTRs and just define gene order based on the coding exons. If a gene is nested in an intron between two coding exons of another gene then we describe that as nesting. So in this example, Raf is upstream of Ilp6
The Genome Browser Gateway should default to the correct assembly once you click on the Drosophila species in the left-hand table. To double check, you are using the correct one, you can see which assembly you should be using via the “Genome Browsers” column of the Pathways Project Genome Assemblies web page. 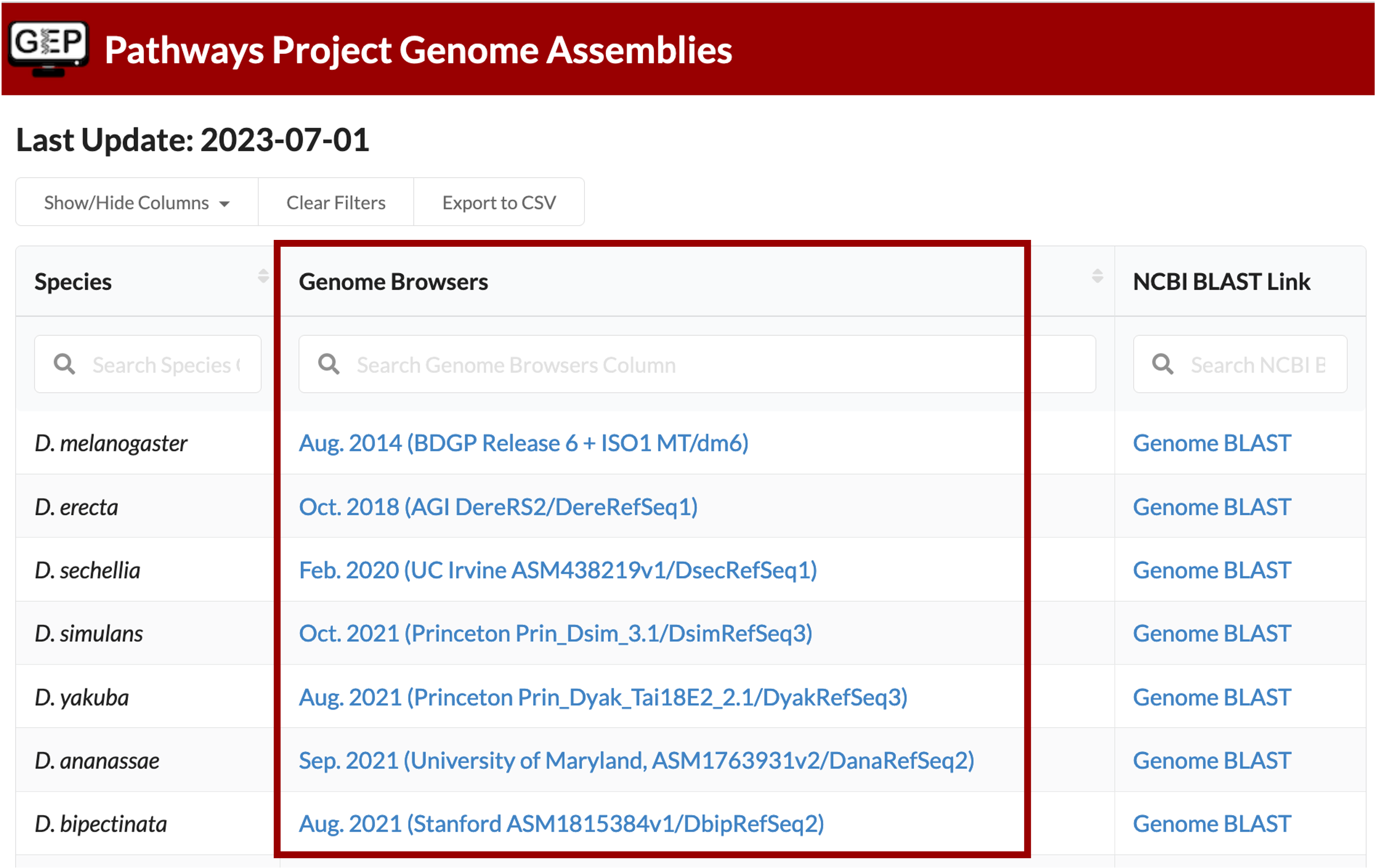
For example, D. yakuba has three assembly options to choose from and according to the Genome Assemblies page, we should use the “Aug. 2021 (Princeton Prin_Dyak_Tai18E2_2.1/ DyakRefSeq3)” assembly when annotating D. yakuba.