 If you’re anything like me, you’ve been doing a lot of puzzles during the COVID-19 pandemic. It is a genuinely satisfying feeling to start with a box full of seemingly unrelated pieces and end up with a complete image of an Italian countryside, outer space, or your favorite cartoon character. There are many types of puzzles, varying in difficulty, size, and number of pieces.
If you’re anything like me, you’ve been doing a lot of puzzles during the COVID-19 pandemic. It is a genuinely satisfying feeling to start with a box full of seemingly unrelated pieces and end up with a complete image of an Italian countryside, outer space, or your favorite cartoon character. There are many types of puzzles, varying in difficulty, size, and number of pieces.
Many of us encounter other types of puzzles in our daily lives—for example, computer scientists and programmers solve mathematical puzzles, engineers solve mechanical problems, and lawyers solve logic problems.
For biologists, one of the biggest puzzles is DNA sequencing, which is surprisingly similar to putting together a jigsaw puzzle. And like jigsaw puzzles, there are many types of sequencing that scientists use based upon the problem they are trying to solve. We will discuss some of them here.
 DNA contains instructions for living things to develop, grow, and function. Variations in DNA can make a difference in how a living thing looks, develops, or responds to its environment. Take humans for example. Humans are 99.9% genetically identical, yet we also have variation in our traits. While some of the differences between each of us comes from our environment and life experiences, our DNA plays an important role in determining our appearance, our traits, and our health.
DNA contains instructions for living things to develop, grow, and function. Variations in DNA can make a difference in how a living thing looks, develops, or responds to its environment. Take humans for example. Humans are 99.9% genetically identical, yet we also have variation in our traits. While some of the differences between each of us comes from our environment and life experiences, our DNA plays an important role in determining our appearance, our traits, and our health.
Being able to analyze an individual’s DNA and detect variation is important for answering questions about health, disease, and other biological phenomena. Scientists use a technology called DNA sequencing to study and analyze DNA.
DNA sequencing is the name given to any process where scientists determine the order of the four chemical building blocks, called bases, that make up the DNA molecule. The four bases are adenine (A), thymine (T), cytosine (C), and guanine (G). DNA sequences help scientists understand the type of genetic information that is carried in the DNA, which may affect its function in the body.
The Human Genome Project used a sequencing method called Sanger sequencing to determine the first near-complete human genome. Sanger sequencing, also called the chain-termination method, was the most widely used sequencing method from the late-1970’s to the early-2000’s. The method is based upon the natural method of DNA replication, the process by which DNA is copied by an enzyme called DNA polymerase.
During DNA replication, DNA unzips its double-helix into two single strands then polymerase binds to the single-stranded DNA and adds new bases, one at a time. As it adds the bases, it fills in the single-stranded DNA to be a full, double-stranded DNA molecule.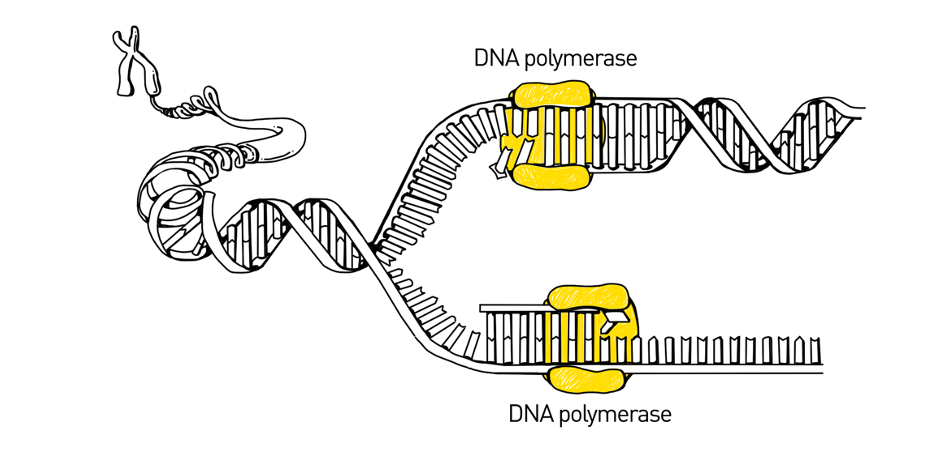
Sanger sequencing takes the concept of DNA replication and uses it to read the sequence of the DNA. The reaction includes special bases called dideoxyribonucleotide triphosphate (ddNTPs) that are randomly incorporated by DNA polymerase into the strand it is copying. These special bases stop DNA polymerase from adding any more bases, resulting in different sized pieces of DNA. The process is repeated over and over, leaving scientists with a bunch of different sized pieces of DNA.
Scientists then sort the fragments by size, and read out the sequence of the bases based on the presence of a dye-molecule attached to the special bases at the end of each fragment. For example, if the shortest fragment ended in a special “A” base, the scientists know the first base in the sequence is adenine. In this way, the scientists build a DNA sequence base by base.
Although the technique has been automated, it is still slow because only one sequence can be read at a time, making it expensive from a labor and materials standpoint. Although newer, automated sequencing technology is more frequently used today, Sanger sequencing still remains a gold standard method and is widely used for smaller-scale projects, and for validating results from other sequencing methods.
![]() Following the monumental completion of the first human genome sequence, researchers started to demand techniques that would enable them to complete sequencing more rapidly and at a cheaper cost. This led to the development of high-throughput, next-generation sequencing platforms. Next-generation sequencing is much faster and cheaper than Sanger sequencing. For example, the first human genome took thirteen years and cost an estimated $2.7 billion to complete. Today, a human genome can be sequenced in a day for a fraction of the cost of the first human genome.
Following the monumental completion of the first human genome sequence, researchers started to demand techniques that would enable them to complete sequencing more rapidly and at a cheaper cost. This led to the development of high-throughput, next-generation sequencing platforms. Next-generation sequencing is much faster and cheaper than Sanger sequencing. For example, the first human genome took thirteen years and cost an estimated $2.7 billion to complete. Today, a human genome can be sequenced in a day for a fraction of the cost of the first human genome.
The principles of next-generation sequencing are similar to Sanger sequencing in that the bases of a small section of DNA are identified and recorded using dye-labeled nucleotides added by DNA polymerase. However, rather than being limited to just a few DNA fragments, next-generation sequencing extends this process so that millions of samples can be sequenced, all at the same time.
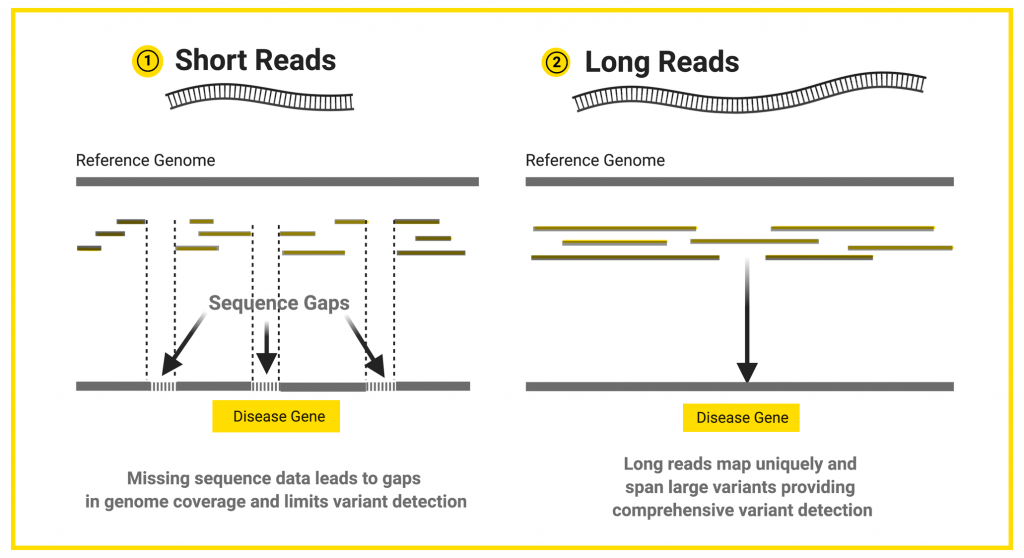
Next-generation sequencing is performed by generating millions of “short” sequences, called reads, which are generally around 150 base pairs long. If the researcher is studying a whole genome, these short-reads are pieced back together like a puzzle using a reference genome as a template. A reference genome is a representative example of a set of chromosomes for a species, produced from the DNA of one or more members of that species. Genetic variants are detected by comparing the puzzle pieces to the reference genome to look for variations in the sequences.
Next-generation sequencing is cost-effective, accurate, and supported by a wide range of analysis tools and pipelines. Short reads perform well for the identification of single base variants and small insertion and deletions, but they are not well suited to the detection of larger sequence changes such as insertions, deletions, duplications, inversions, or translocations that affect more than fifty bases.