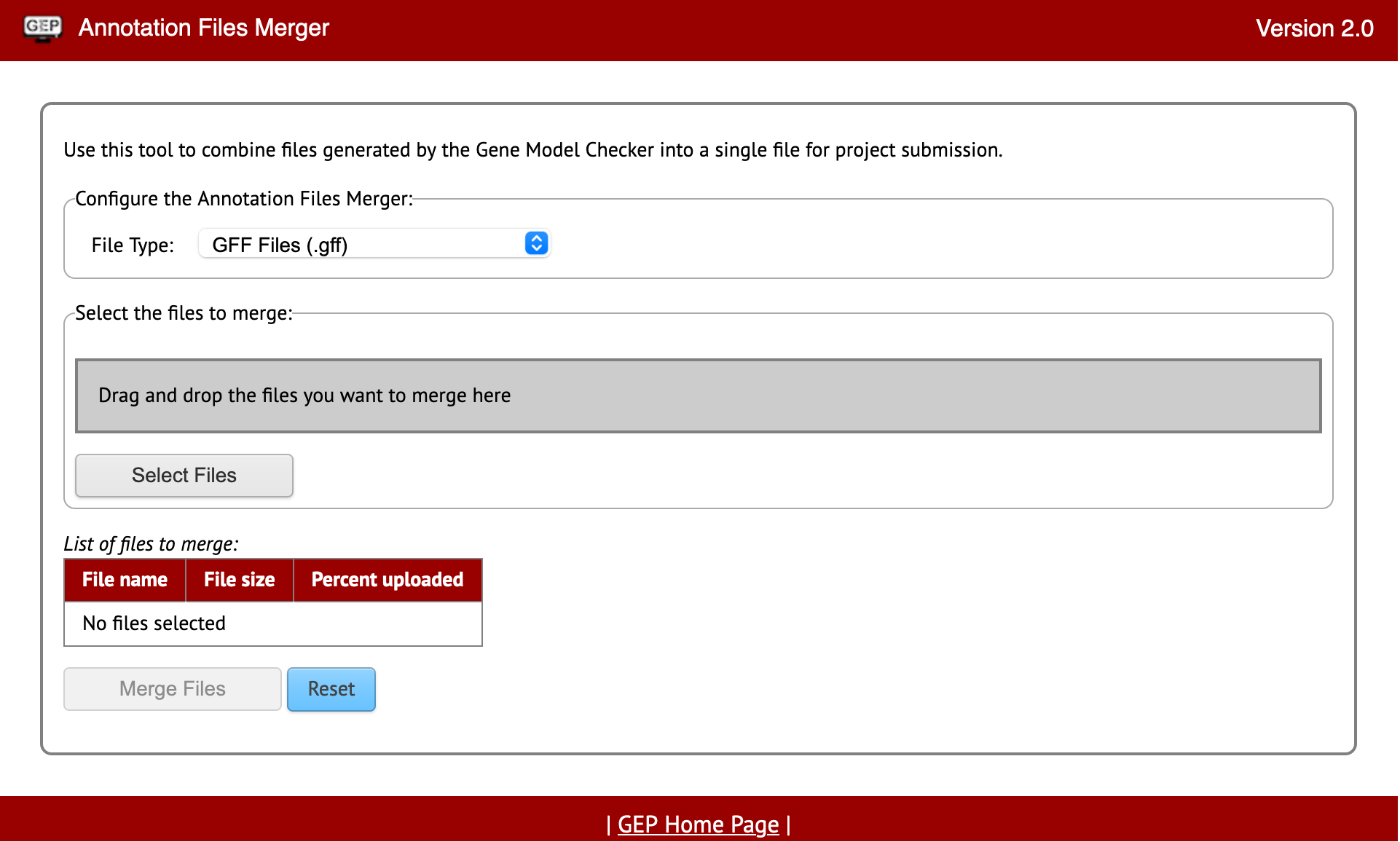
 The Annotation Files Merger allows for the merging of multiple files gathered during the annotation process. This is important because it allows consistency in data collection and a quick method to review everything from a top-down glance. The supported file types for the annotation files merger are: GFF (Generic Feature Format), FASTA (FAST-ALL), PEP (Peptide File Format), and VCF (Variant Call Format).
The Annotation Files Merger allows for the merging of multiple files gathered during the annotation process. This is important because it allows consistency in data collection and a quick method to review everything from a top-down glance. The supported file types for the annotation files merger are: GFF (Generic Feature Format), FASTA (FAST-ALL), PEP (Peptide File Format), and VCF (Variant Call Format).
The file types listed will be found throughout your annotations so it will also be important to recognize what files are which and why they are important too.
Reminders
- Even if the isoforms are identical, you should merge each file type for every isoform:
- GFF (Generic Feature Format): located in the Gene Model Checker and provides a soft overview of your genomic annotation
- FASTA (FAST-ALL): found in the Gene Model Checker; provides the nucleotide sequences that comprise your annotation
- PEP (Peptide): also found in the Gene Model Checker; provides the same information as a .FASTA file but in the format of peptides vs. nucleotides
- VCF (Variant Call Format): seldomly used but very important when finding consensus errors; provides information to the Gene Model Checker and our models to update a scaffold with the most accurate readings
- Most Importantly: You do NOT need to open or edit these files. Do NOT do this. These files, if edited, will actually change the research you spent much time compiling and completing. Just download the file.
See the Annotation Files Merger User Guide for more information.
 FlyBase is a bioinformatic database for all things Drosophila. This website can be a great place to study the who, what, where, when, and why behind a particular gene within D. melanogaster. You can best use FlyBase by searching for individual genes to uncover detailed reports that summarize genomic location, functionality, orthological pairing, and much, much more.
FlyBase is a bioinformatic database for all things Drosophila. This website can be a great place to study the who, what, where, when, and why behind a particular gene within D. melanogaster. You can best use FlyBase by searching for individual genes to uncover detailed reports that summarize genomic location, functionality, orthological pairing, and much, much more.
Reminders
- If you are looking for a deeper understanding of your assigned genes – look no further than FlyBase.
- It can be incredibly dense within the FlyBase reports, so be sure to have a general idea of what you are looking for.
- When looking for a gene, remember that gene names are case-sensitive.
See the FlyBase Tools and Downloads Documentation for more information.
The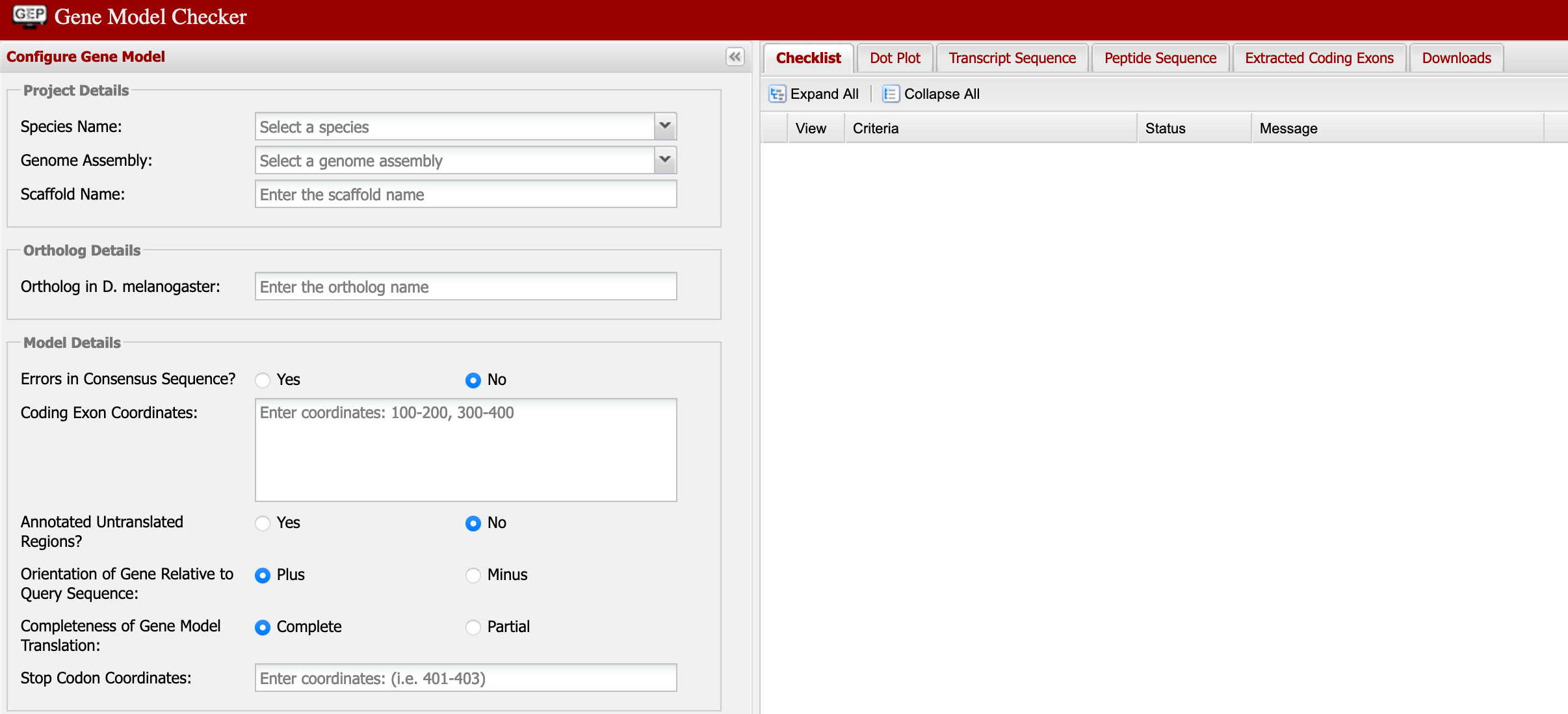 Gene Model Checker is a key “checkpoint” in the annotation process. This allows us to visualize our annotation as compared to that of the D. melanogaster gene. Here we can see a bigger picture as to what may or may not be missing so be sure to review your dot plot and protein alignment for possible errors or common mistakes.
Gene Model Checker is a key “checkpoint” in the annotation process. This allows us to visualize our annotation as compared to that of the D. melanogaster gene. Here we can see a bigger picture as to what may or may not be missing so be sure to review your dot plot and protein alignment for possible errors or common mistakes.
Reminders
- The Gene Model Checker is where you will find:
- Dot Plots for visualization
- Protein Alignments from your target vs. D. melanogaster
- Be sure to download all of your PEP, FASTA, and GFF files here too. Don’t skip any of them.
See the Gene Model Checker User Guide and Video Tutorial for more information.
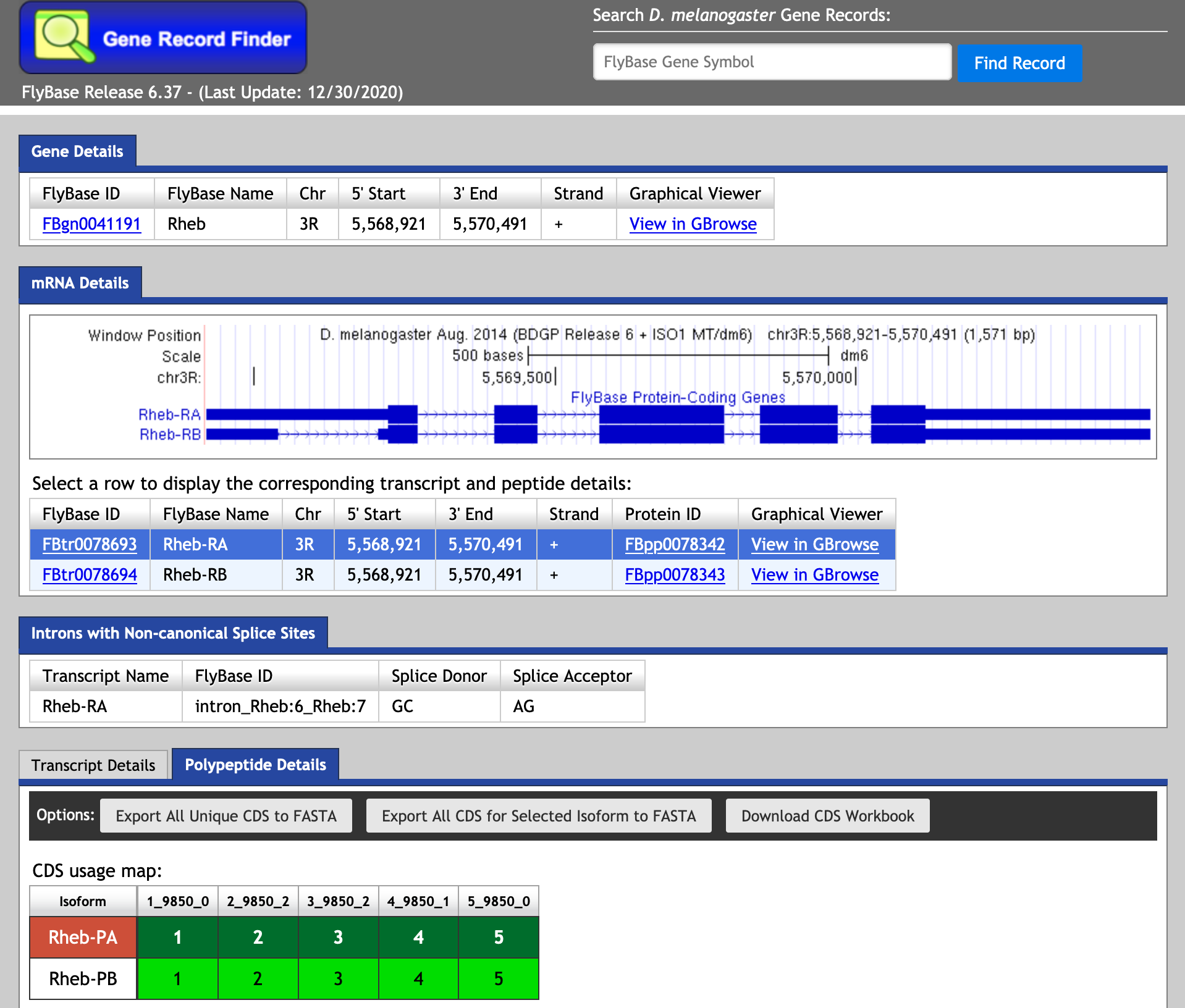 The Gene Record Finder can be used to break down the complex information from FlyBase into a quick and easy-to-read form. More importantly, the Gene Record Finder can provide transcription details, polypeptide details, and isoforms for the gene you are examining.
The Gene Record Finder can be used to break down the complex information from FlyBase into a quick and easy-to-read form. More importantly, the Gene Record Finder can provide transcription details, polypeptide details, and isoforms for the gene you are examining.
Reminders
- If you are annotating the UnTranslated Regions (UTRs), be sure to use the Transcription Details.
- If you are looking for gene CoDing Sequences (CDSs), be sure to use the Polypeptide Details.
- Make sure you account for EACH isoform. Unique or identical, we need information on all isoforms!
See the Gene Record Finder User Guide for more information.
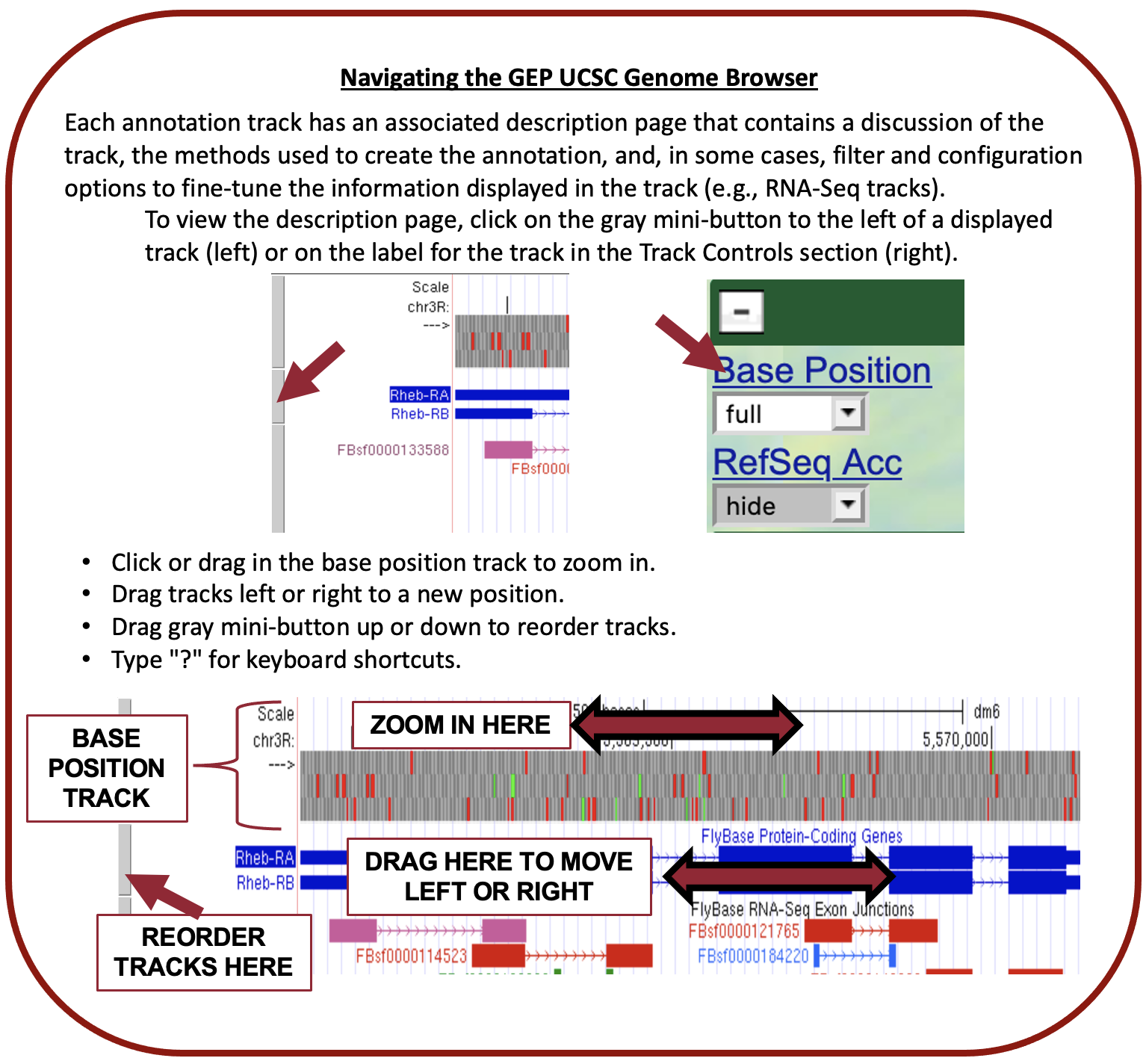
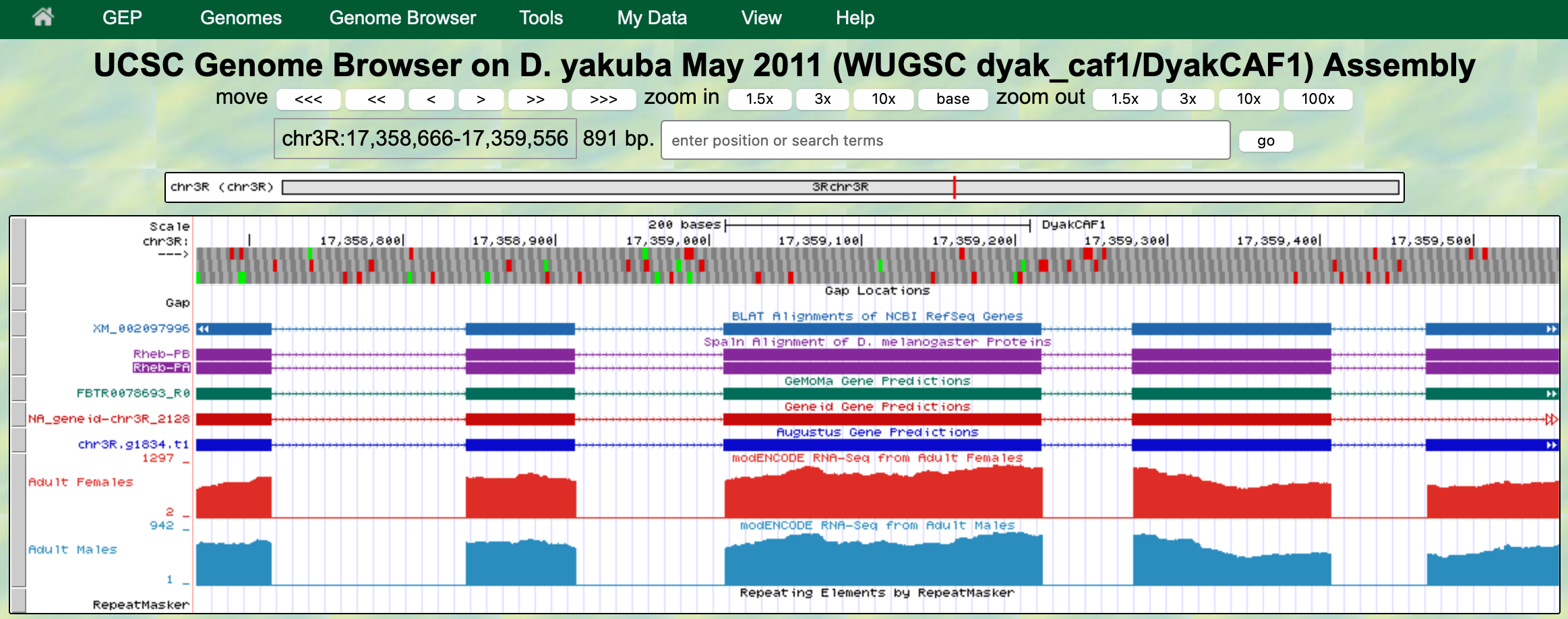 The GEP UCSC Genome Browser is a tool to visualize genetic information in an easy-to-use format. This makes it far easier for us to examine unique patterns or spot similarities across species.
The GEP UCSC Genome Browser is a tool to visualize genetic information in an easy-to-use format. This makes it far easier for us to examine unique patterns or spot similarities across species.
Reminders
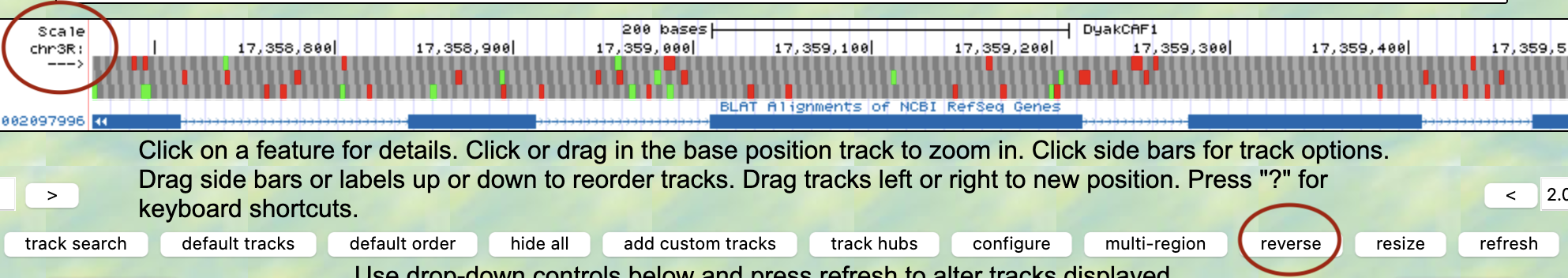
- Be sure to check if your gene is located in the reverse or forward position in relation to your scaffold. This error commonly occurs!
- This can be fixed by clicking the “reverse” button (located below your reference image) or the “arrow” located on the far left-hand side.
- Methods to check this can be found with in-frame stop codons and a missing Methionine in CDS1.
- Dependent on gene direction, remember that if the
- gene is right to left: Downstream ← Gene ← Upstream
- gene is left to right: Upstream → Gene → Downstream
- Remember your BLAST results and compare them to each CDS. BLAST will not provide a perfect match each time so use it as a guide and not as an absolute metric.
See the UEG | Genome Browser Video for a breakdown of how to use the GEP UCSC Genome Browser.
 BLAST is vital for success in genomic annotation as a whole. As mentioned in the introductory video for navigating and interpreting BLAST results, there are numerous steps that need to be done in order to make sure BLAST is working for you and you aren’t working for BLAST.
BLAST is vital for success in genomic annotation as a whole. As mentioned in the introductory video for navigating and interpreting BLAST results, there are numerous steps that need to be done in order to make sure BLAST is working for you and you aren’t working for BLAST.
Reminders
- BLAST is heuristic and deterministic, which means it will not give you the same answer every single time, so it should only be used as a guide, along with other lines of evidence for your model.
- Evidence gathered from BLAST is not always exact or precise. Be sure to check your coordinates carefully!
- Within BLAST be sure to collect these key points from the top two hits:
- Accession Numbers
- E-values
- Percent Identity
- Frame of Reference and Strand (+/-)
- Approximate coordinates of the start and stop for each CDS
To ensure you are using the correct type of BLAST, consider the following:
| BLAST Type | Query (sequence to match) | Database/Subject (searching for match) | Function | Use Cases |
|---|---|---|---|---|
| blastn (nucleotide) | nucleotide | nucleotide | searching with shorter queries, cross-species comparison | map mRNAs against genomic assemblies |
| blastp (protein) | protein | protein | general sequence identification and similarity searches | search for proteins similar to predicted genes |
| blastx | nucleotide → protein | protein | identifying potential protein products encoded by a nucleotide query | map proteins/CDS against genomic sequence |
| tblastn | protein | nucleotide → protein | identifying database sequences encoding proteins similar to query | map proteins against genomic assemblies |
| tblastx | nucleotide → protein | nucleotide → protein | identifying nucleotide sequences similar to the query based on their coding potential | identify genes in unannotated sequences |
Arrows indicate the BLAST program translates the nucleotide sequence before performing the search.
See the Introduction to NCBI BLAST and Introduction to BLAST using Human Leptin lessons for more information.
The Pathways Project Genome Assemblies page is by far the quickest and most effective way to navigate to BLAST if you are looking to search against a specific assembly.
Reminders
- Pathways Project Genome Assemblies searches against SPECIFIC species assemblies. If you are looking for something a bit broader, you can navigate to NCBI BLAST.
The Sequence Updater creates a VCF (Variant Call Format) file, which can be used to update an existing assembly. This is used whenever a student may have sufficient evidence to suggest that an assembly has an error causing an incorrect alignment.
Reminders
- Before suggesting a consensus error, make sure to contact your instructor and/or a TA to confirm that you have looked at all other lines of evidence; using the Sequence updater and proposing a consensus error should always be your last resort. Updating a sequence does not happen often and should not be used unless there is significant evidence to suggest it. More often than not, this tool will not be used.
- You must have multiple sequences that suggest an error. Much like verifying your work, it’s better to have numerous pieces pointing to the same potential error than to only have one. Is the change you are seeking valid and accurate?
- As outlined in the GEP Tools | Sequence Updater video tutorial, keep in mind once you apply these changes, that’s it. Be absolutely certain this is what you want.
Do you have an exon that is too small to find with BLAST? Is the result, no matter what you change, coming up with “No Significant Matches Found?” If so, the Small Exons Finder can offer some insight in locating small exons that would otherwise be finding a needle in a very large haystack.
Reminders
- When using the Small Exons Finder, you should use the whole scaffold from the GEP UCSC Genome Browser and not just the position of your predicted gene. You can do this by following the steps below:
- Navigate to VIEW > DNA
- Remove the coordinates and the “:” from position, which will leave only the scaffold name, and then select > get DNA
- Save this output in its entirety as a .FASTA format (e.g., example_name.FASTA)
- Before searching, double check your search parameters, specifically, make sure that your subrange is appropriate, else you will get a lot of matches.
If you bump into a situation where your gene annotation contains many in-frame stop codons or even if your initial CDS doesn’t appear to have a start Methionine, more than likely, you are examining your gene in the wrong orientation.
- Your BLAST results should indicate if your query is expected to be on the positive or negative strand. Be sure to read your BLAST report thoroughly.
- If your gene is on the negative strand, click on the “reverse” button to flip the gene’s orientation so it still reads from left-to-right. The figure below shows what this might look like when examining your first and second CDSs.
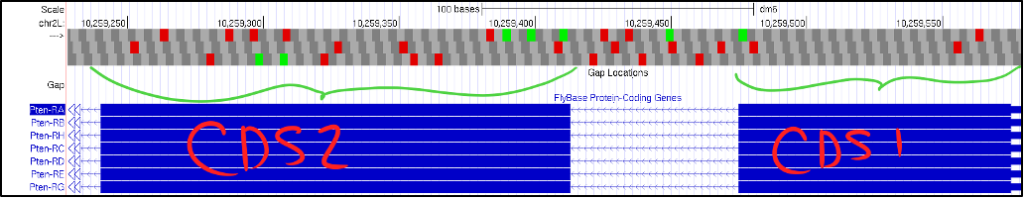
- Notice in the picture above of CDS1 and CDS 2, we see two crucial errors in our reading:
- We do not see a start Methionine (ATG) in CDS1.
- Each frame in CDS2 has multiple in-frame stop codons.
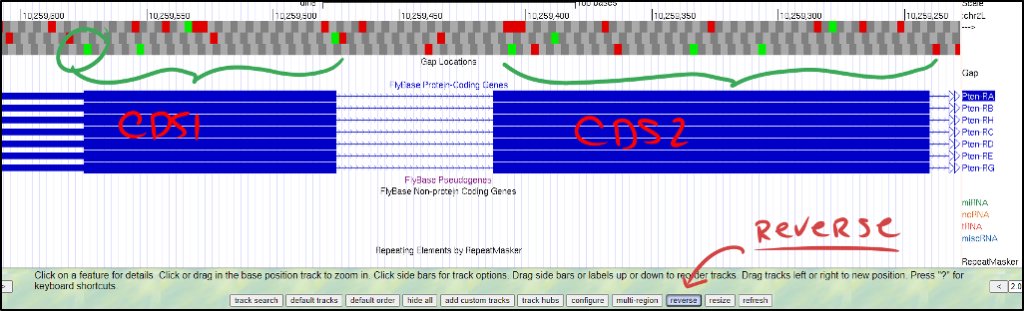
It’s okay if the gene is on opposite strands in D. melanogaster and your ortholog. Ensure the rest of the genomic neighborhood makes sense (i.e., adjacent genes on opposite strands in D. melanogaster should also be on opposite strands in your ortholog). In an ideal world, the relative orientation and placement of genes should be consistent.
It depends. Tracks have been designed within the GEP UCSC Genome Browser based on the kind of research being done. It is essential to ask the question, “What information am I looking for that the Genome Browser can provide to me?” Examples could be RNA-Seq data, in-frame stop codons, comparative genomics across a genus, or many other things. In general, though, we recommend selecting your “Default Tracks” to get started. Aside from this, we have several other tracks that will stand out during your annotation work, see below:
- Mapping and Sequencing Tracks → Base Position → FULL
- Genes and Gene Prediction Tracks → FlyBase Genes → PACK
- RNA Seq Tracks → FlyBase Exon Junctions → PACK
- Updated Transcriptome Tracks → Splice Junctions (discretionary) → PACK
- Comparative Genomics tracks (if available)
At first glance, it can be challenging to know what nucleotide position the GEP UCSC Genome Browser is showing you. A useful method of reading this frame can be found by looking for the pipe, denoted with this “|” symbol. By looking for the number in between each pipe, you can quickly figure out which nucleotide corresponds with the overall scaffold count.
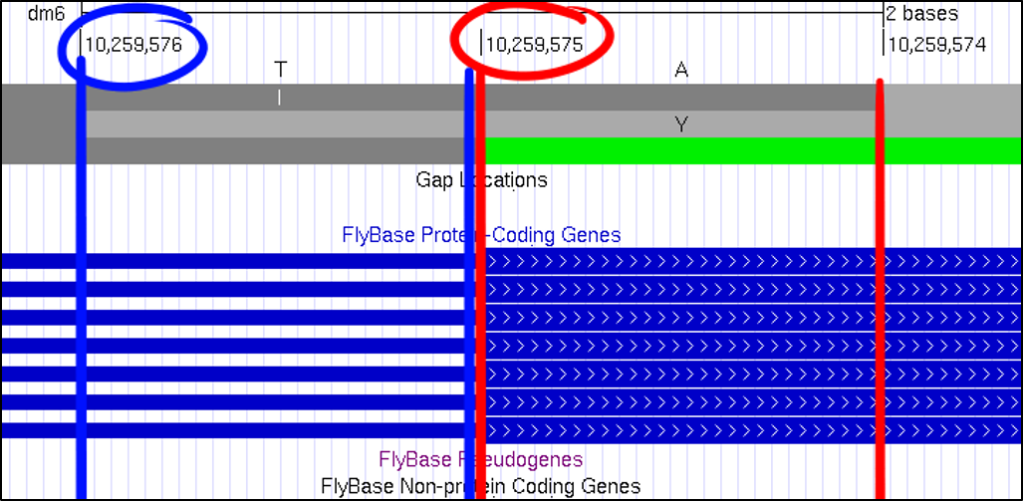
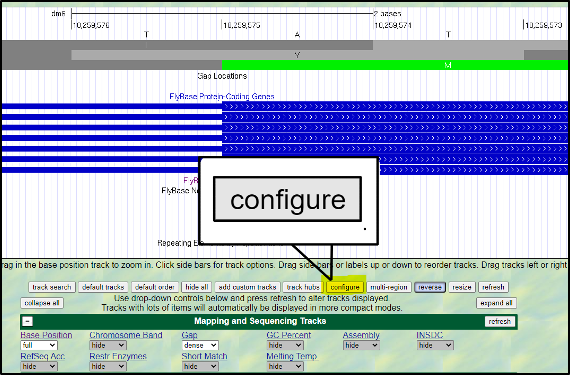
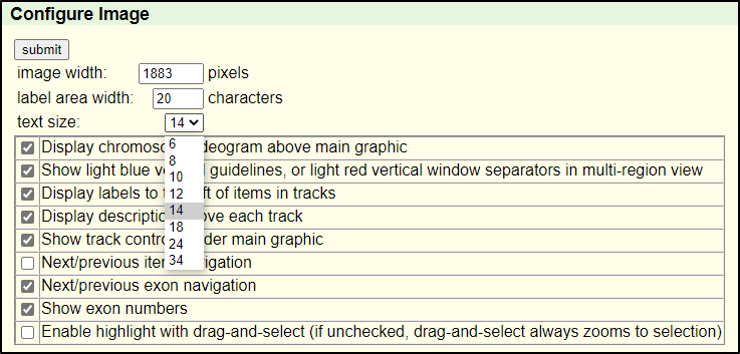
Absolutely! You can increase the font size by clicking on “configure” and then selecting a different “text size.” This font size can be adjusted to many different sizes – so pick whichever you find most comfortable.
BLAST, known as the Basic Local Alignment Search Tool, can identify similarities or differences within biological data. It accomplishes this by providing the user an E-value (Expected Value) to determine statistical significance. Explanations of each of the five types of BLAST and their uses can be found below:
| BLAST Type | Query (sequence to match) | Database/Subject (searching for match) | Function | Use Cases |
|---|---|---|---|---|
| blastn (nucleotide) | nucleotide | nucleotide | searching with shorter queries, cross-species comparison | map mRNAs against genomic assemblies |
| blastp (protein) | protein | protein | general sequence identification and similarity searches | search for proteins similar to predicted genes |
| blastx | nucleotide → protein | protein | identifying potential protein products encoded by a nucleotide query | map proteins/CDS against genomic sequence |
| tblastn | protein | nucleotide → protein | identifying database sequences encoding proteins similar to query | map proteins against genomic assemblies |
| tblastx | nucleotide → protein | nucleotide → protein | identifying nucleotide sequences similar to the query based on their coding potential | identify genes in unannotated sequences |
Arrows indicate the BLAST program translates the nucleotide sequence before performing the search.
Each BLAST tool has a different function. You should understand that various tools of BLAST will provide you with different outcomes. If you use the wrong tool, you should expect information that doesn’t make sense and precious time lost. See the table below to see examples of how each tool of BLAST can be used in relation to your time with the GEP. If you ever see results that do not make sense, feel free to reach out to the GEP TA’s, and we will be more than happy to provide more in-depth explanations and assistance with your queries.
| BLAST Type | Description of Usage as a Scientific Question |
|---|---|
| blastn | “Are there nucleotide similarities from D. melanogaster to D. yakuba?” |
| blastp | “Are there peptide similarities from D. melanogaster to D. yakuba?” |
| blastx | “Are there peptide similarities inside of D. melanogaster that I can find with only my nucleotide sequence from D. yakuba?” |
| tblastn | “Are there nucleotide similarities inside of D. melanogaster that I can find with only my peptide sequence from D. yakuba?” |
| tblastx | “Are there translated nucleotide similarities from D. melanogaster that are found in the translated nucleotides of D. yakuba?” |
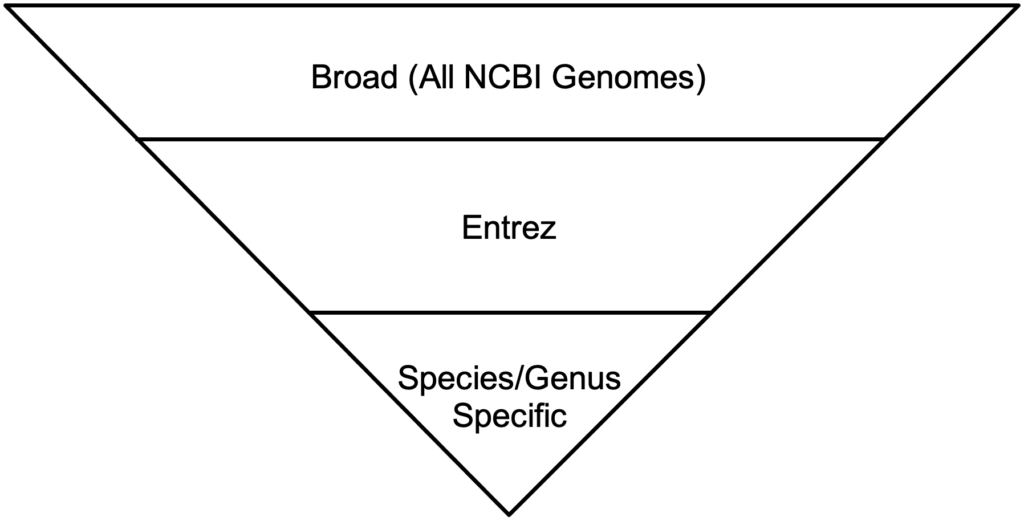
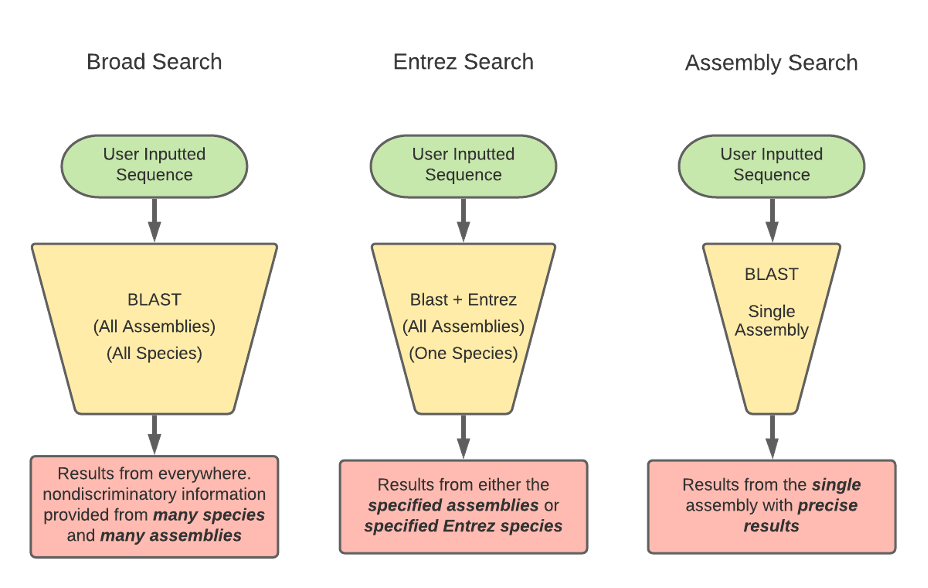
BLAST has three methods for the GEP to understand a target species and our reference species D. melanogaster. Instead of searching broad areas (all NCBI genomes) that may have no information pertaining to your query, BLAST can also be used for more narrow searches such as for Entrez and Assembly searches. It is easy to understand these different search queries as an overlapping funnel that varies based on what you are searching for with BLAST. The figures below show each query type and how they relate to the search results.
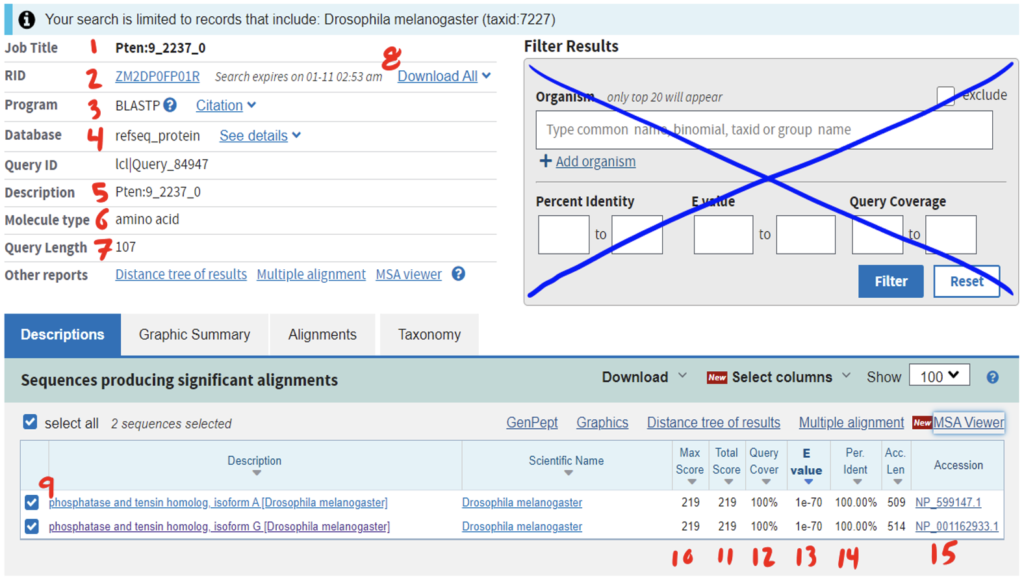
Reading the BLAST results page can be daunting at first. Be sure to anchor yourself to your initial scientific question so you don’t get lost. For a detailed breakdown of reading BLAST results, refer to the GEP Tools | NCBI BLAST video tutorial. Below is an example of a BLAST results page and a legend with specific explanations for how to interpret the page.

The RefSeq protein track found in the GEP UCSC Genome Browser is linked to RefSeq: NCBI Reference Sequence Database. When given a REFSEQ protein track, we need to ask the questions, “What is the predicted function of this gene?” and “Does this predictive function match what is found in D. melanogaster?” This, in turn, will give us a better scope of if we have located our target D. melanogaster homolog.
With a peptide sequence or even the Protein ID, we will need to use blastp. The tricky part of searching for a predictive function is that we don’t want to search for everything since this will take far too long and render many useless results. Consider the following questions:
- “What kind of input x and output f(x) am I expecting?”
- Example: “I have a Protein sequence and am looking for Protein returns.”
- “Do I need results from hits outside of my genus, within the genus, or a specific species within my genus?”
- Example: “I have a Protein sequence from D. kikkawai, and I am trying to find its predictive function from D. melanogaster.”
Assuming the answers to our questions above are “blastp” and “within the genus,” we can run this report by calling on an Entrez search with D. melanogaster (taxid: 2772).
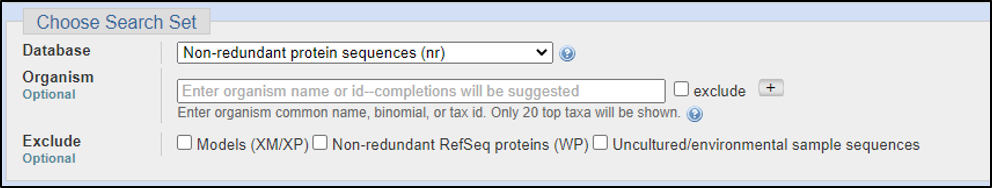
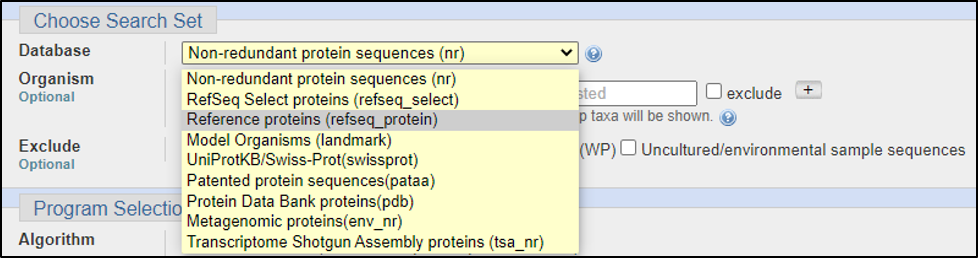
BLAST relies on statistical analysis to determine areas between inputs and databases to produce statistically significant results. Sometimes when inputting a specific coding DNA sequence (CDS) or query, it is possible that BLAST will not warrant any results. This can happen for numerous reasons, three of the most common are listed below:
- No significant results are found. As simple as it sounds, sometimes, through enough evolution, CDSs can drastically change from one species to the next. If your species is several steps away from D. melanogaster, this CDS may be drastically different.
- NOTE: When examining this kind of change, you can use the “Comparative Genomic” tabs in the GEP UCSC Genome Browser to provide evidence of this. Don’t expect to jump to this conclusion immediately – we are scientists and want to see why we are getting this result.
- The query submitted may not have been submitted correctly. Be sure to look back at which BLAST tool you are using and exam the query sequence you are comparing to it.
- Sometimes the information you are searching for can be “lost in translation.” This can cause problems with several search parameters, and BLAST ends up providing too little or too much and simply doesn’t want to flood you with information. To fix this kind of error, we suggest refining your search. A further explanation of refining your CDS can be found in the GEP | Pathways Annotation – CDS Annotation and Refinement video.
The short answer is yes, BLAST is essential. Many computational biologists, geneticists, and numerous other scientists use BLAST day-to-day for complex calculations and analysis. Without BLAST, we would not be able to accomplish most of what is done during Bioinformatic analysis. Could you imagine having to calculate these E-values and Alignments by hand? It isn’t fun. Remember that BLAST is a tool, and like all tools across industries, it is better to stay ahead of the curve and be confident in your ability to use this widely demanded tool.
An e-value, also known as an expected value, is a value provided by BLAST to denote expected random chance when searching the database with your query sequence. The closer your e-value to zero, the less noise we expect to see and the more “significant” our match becomes. Just because we get an e-value of 0.0, doesn’t mean it is perfect though. Let’s examine two situations where an e-value would be 0.0 for different reasons:
- From BLAST, an e-value of 0.0 can occur when we examine short sequences against other short databases that are near identical. According to NCBI, “shorter sequences have a higher probability of occurring in the database purely by chance.” As scientists and annotators, we do not look for chance, we look for evidence to form solid hypotheses and ideas.
- From a computer science perspective, systems hold values as integers and floating point numbers (has a decimal point). When dividing or calculating chances and limits, these floating points, much like on a calculator, have a set amount of space. Because all computers contain what is known as a “Floating-Point Error,” BLAST mitigates this error by rounding to an e-value of 0.0 when our value is < 5*10e-324. To put that into perspective, 5*10e-324 looks like this:
0.00000000000000000000000000000000000000000000000000000000000000000000000 0000000000000000000000000000000000000000000000000000000000000000000000000 0000000000000000000000000000000000000000000000000000000000000000000000000 0000000000000000000000000000000000000000000000000000000000000000000000000 00000000000000000000000000000000005.
- Simply put, it is because our design of computers in bits is in a Log2 format, this kind of floating-point error is here to stay for a while.
- Overall, e-values that approach 0.0 or 0e0 are far lower than e-values approaching 1. Always think in terms of “noise.” The less noise, the better and more confident we can be in our results.
BLAST results within the GEP should not take several hours. Based on our query sizes, you should expect search times of between seconds to several minutes only. If you are getting search times of several hours, we have a few suggestions for this:
- Review what query sequence you have submitted and against what database.
- If the incorrect database or query is submitted, BLAST can take much longer to fulfill your request. Take a moment to review your query and databases and run them again.
- Peak Times occur within BLAST. It is best to run a BLAST search on a different internet browser tab at specific peak times. This is due to BLAST being a cloud computing service used by thousands of scientists daily. Being one of those scientists, you will experience the very same delay.
- These peak times vary, but BLAST will indicate when they occur.
For more information on troubleshooting your BLAST results and why these results may be taking so long, feel free to reach out to any of the GEP Virtual TAs or see NCBI’s troubleshooting page.
In the Pathways Project, you will need to keep critical pieces of information on standby. There will be times where you will not finish an annotation or will need to navigate through multiple tabs at any given time. Document the following for quicker navigation:
- Target Gene’s Scaffold and its accession number
- Target Gene’s genomic coordinates
- Pathways Project: Annotation Form
The Gene Record Finder uses FlyBase to collect the most pertinent information you will need during your time annotating your target gene. Because of how this webpage interacts with the FlyBase records, you must type the target gene symbol precisely as indicated, as they are case-sensitive. Even if one capital letter, one dash, or even a single character isn’t exactly as it is found on FlyBase, you might get an error that looks like the figure below: